In this step, you will use PCR to amplify the DNA barcode for your samples. This will take about 3 hours, but most of it will be waiting time. The primer pair you use will depend whether you have sampled an animal, bird, fungus, or plant:
- LCO1490/HCO2198 for animals
- Bird F1/Bird R1 for birds
- ITS1F/ITS4 for fungi
- rbcL-1F/rbcL-724R for plants
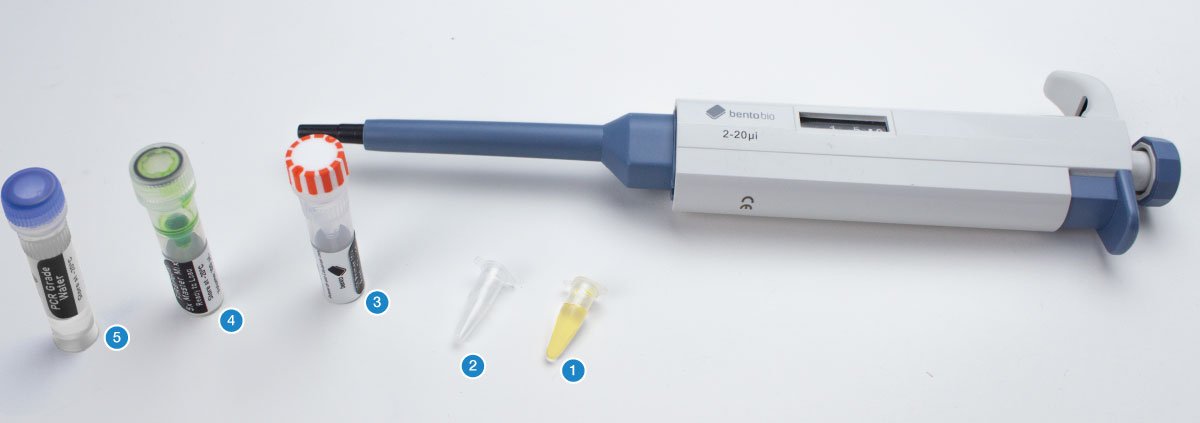
You will need your DNA extractions (1), one empty PCR tube per sample, plus one or two for controls (2), an empty 1.5 mL microcentrifuge tube, the primer pairs for this project (forward and reverse tubes) (3), 5x FIREPol® master mix (4), and PCR grade water (5).
The first step is to calculate how much PCR mix you require for your samples.
PCR mix is the combination of 5x master mix, primers, PCR grade water, and any other additives that you will add to each PCR tube before adding your DNA template. You can think of the 5x master mix as a concentrate that lacks primers, and the PCR mix that you make from it as the complete 1x PCR reagent mix that is ready to amplify extracted DNA.
For each sample, you will need:
- 4 µL of 5x FIREPol® master mix (1/5 of the total PCR volume)
- 13.2 µL of PCR grade water (total volume of the PCR minus everything else)
- 0.4 µL of the forward primer in the primer pair (to give a 0.2 µM final concentration)
- 0.4 µL of the reverse primer in the primer pair (to give a 0.2 µM final concentration)
- A volume allowance for 2 µL of DNA template, which is added later (this volume can be adjusted)
You also need a negative control and optionally a positive control.
A negative control is a PCR tube of PCR reaction mix to which no DNA is added. This should produce no PCR amplicons and is used to check your PCR reaction mix is not contaminated.
A positive PCR control is a PCR of a sample or DNA extract that is known to work. This should always produce a PCR amplicon and is used to confirm that your PCR is working even if your samples fail to amplify. It is not needed if your samples work, but it is extremely useful if your samples fail for any reason.
As you can see from the reagents listed above, there are four reagents that need to be added to each PCR mix, and some of those volumes are too small to pipette using a 20 µL pipette. This means that you need to use a batch PCR mix containing shared reagents for all samples and controls, and then aliquot (divide the solution) into individual PCR tubes. This batch approach can also save time, pipetting steps, and pipette tips. However, it is a little more complicated to calculate than just preparing a single tube at a time.
To calculate the volumes needed for a batch PCR mix, first calculate the volumes of reagents needed for a single PCR mix, and multiply these volumes by the number of samples plus controls. Then add an additional 10% to account for any pipetting errors (i.e. multiply by 1.1).
Batch reagent volume = PCR reagent volume x (no. samples + controls) x 1.1
For example, if you have 8 samples from your DNA extractions:
8 DNA extractions + 1 negative control + 1 positive control x 1.1 = 11 repeats of PCR reagents
- 11 x 4 µL = 44 µL of FIREPol® master mix
- 11 x 10 µL = 145.2 µL of PCR grade water
- 11 x 0.4 µL = 4.4 µL of forward primer
- 11 x 0.4 µL = 4.4 µL of reverse primer
In this example, you would use the 20-200 µL adjustable pipette to transfer the 44 µL of 5x FIREPol® master mix, 145.2 µL of PCR grade water, 4.4 µL of the forward primer, and 4.4 µL of the reverse primer, into a 1.5 mL microcentrifuge tube. Round any volumes that you can’t pipette exactly to the nearest pipettable volume.
Wear gloves to protect your reagents from contamination. Mix each tube before use. Make sure to use a fresh pipette tip each time.
Close the lid of the 1.5 mL microcentrifuge tube and invert several times to ensure thorough mixing of your PCR reaction mix. Holding the tube between thumb and two fingers, use a flick of the wrist to ensure the PCR mix is all at the bottom.
This creates a batch PCR mix with a total volume of 198 µL. It can be split into 10 PCR tubes of 18 µL (total volume 20 µL when the DNA template is added), leaving a small amount of excess PCR mix in case of pipetting errors.
To make each individual PCR, set the 2-20 µL adjustable pipette to 18 µL and transfer 18 µL of PCR reaction mix into the required number of PCR tubes.
Use a permanent marker to label the PCR tubes with your sample names. Also label the negative control so you know not to add DNA to this PCR tube, and the positive control tube if you are intending to to use one.
Now add extracted DNA to act as a DNA template for the PCR.
If you are using a HotSHOT DNA extraction, set your micropipette to 2 μL. Using a new pipette tip, transfer 2 μL of your DNA extraction into the correspondingly labelled PCR tube containing PCR mix.
Mix briefly by pipetting up or down or stirring. Then discard your tip.
Make sure to keep your DNA extraction upright and pipette from the surface of the liquid.
The DNA extractions contain PCR inhibitors that will prevent your PCR from being successful if the liquid is mixed.
If you are using a Dipstick DNA Extraction, dip the dipstick containing extracted DNA from the sample several times, wipe off any droplets of PCR mix on the inside of the tube, and discard the dipstick.
Once you have transferred the DNA extraction into the PCR tube, close the lid.
Leave the negative PCR control with no added DNA. If you are using a positive control, add the extracted DNA to the positive control PCR tube.
Ensure all the PCR mix is at the bottom of each tube and there are no air bubbles. If there are droplets on the sides or lids, or bubbles, you can force the liquid to the bottom of the tube by holding the tube between thumb and two fingers and use a flick of the wrist to force the PCR mix to the bottom. Alternatively you can tap the PCR tube on a hard surface to do the same.
Place your PCR tubes in the thermocycler block.
Set up the thermocycler using the relevant PCR program for your primer pair:
Note that there are many possible variations of PCR programs for these primer pairs, and some may work better for your particular species than the generic protocols below. The annealing step temperature is particularly important — it can be raised to encourage greater specificity if you encounter non-specific amplification, and lowered to be more tolerant to primer mismatches if PCRs fail.
PCR program for ITS1F/ITS4 primers (fungi):
- Initial denaturing: 15 mins at 95°C
- 35 cycles made of 3 steps
- Denaturing: 30 secs at 95°C
- Annealing: 30 secs at 55°C*
- Extension: 45 secs at 72°C
- Final extension: 10 mins at 72°C
- Store: ∞ at 15°C
Total run-time = 139 mins
PCR program for Bird F1/R1 primers (birds):
- Initial denaturing: 15 mins at 95°C
- 6 cycles made of 3 steps:
- Denaturing: 60 secs at 94°C
- Annealing: 90 secs at 45°C
- Extension: 90 secs at 72°C
- 35 cycles made of 3 steps
- Denaturing: 60 secs at 94°C
- Annealing: 90 secs at 55°C
- Extension: 90 secs at 72°C
- Final extension: 5 mins at 72°C
- Store: ∞ at 15°C
Total run-time = 253 mins
PCR program for LCO1490/HCO2198 primers (animals):
- Initial denaturing: 15 mins at 95°C
- 35 cycles made of 3 steps
- Denaturing: 60 secs at 95°C
- Annealing: 60 secs at 40°C
- Extension: 90 secs at 72°C
- Final extension: 7 mins at 72°C
- Store: ∞ at 15°C
Total run-time = 217 mins
PCR program for rbcL primers (plants):
- Initial denaturing: 15 mins at 95°C
- 35 cycles made of 3 steps
- Denaturing: 30 secs at 94°C
- Annealing: 45 secs at 54°C*
- Extension: 45 secs at 72°C
- Final extension: 5 mins at 72°C
- Store: ∞ at 15°C
Total run-time = 143 mins
(For help setting up a PCR on your Bento Lab visit the PCR Thermocycler User Manual.)
* The primer annealing temperatures listed here are a good starting point based on published studies. Depending on your target organism, you might find that slightly increasing the temperature (to reduce non-specific amplification) or decreasing the temperature (to improve the ability of the primer to bind to the target DNA, especially in the case of mismatches) helps improve your results. If you are working on a specific group of organisms, you can often find recommended annealing temperatures for these primers in published scientific articles about that group.
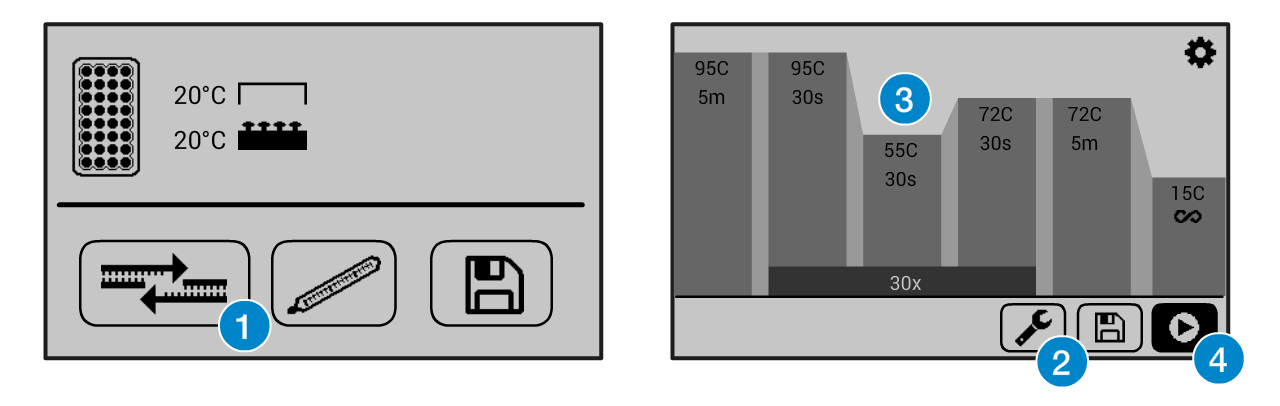
If you need help operating the Bento Lab thermocycler, check the manual. You can use the PCR preset (1), then modify (2) the program to the required settings (3) before running the program (4). This figure shows example settings – please consult the specific protocol for recommended PCR program settings.
The program will run for ca 2 hours. When it is finished, you can keep the result in the freezer, or use it right away for gel electrophoresis.