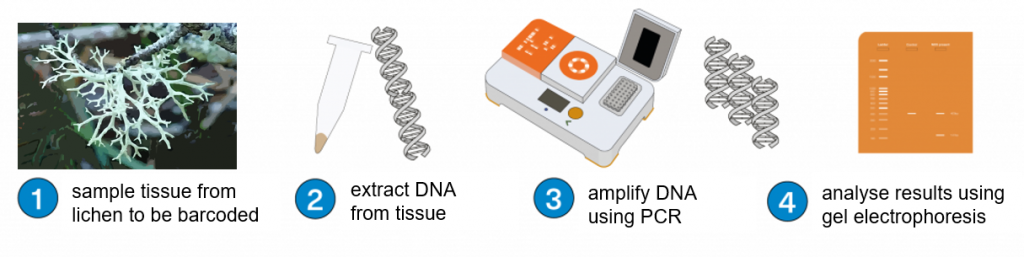
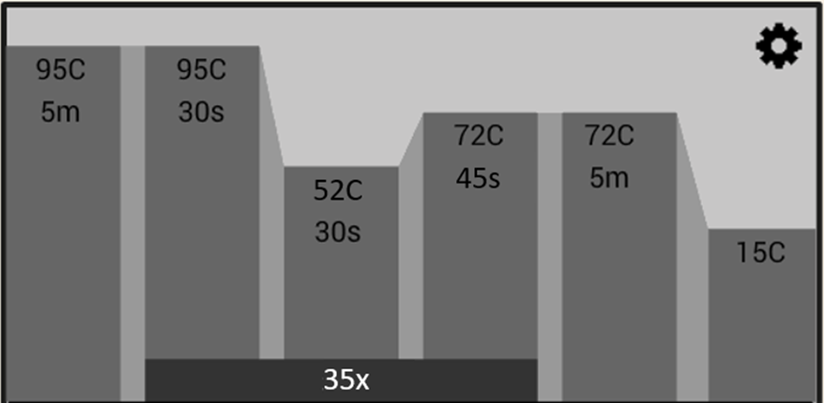
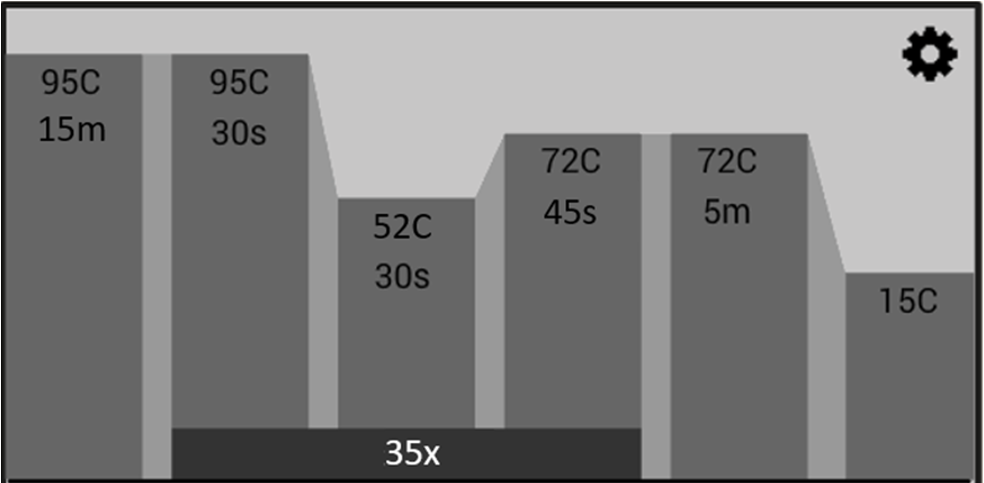
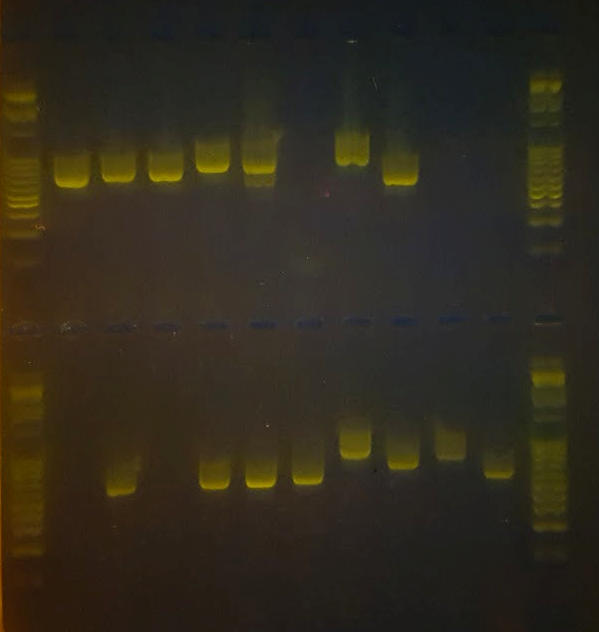
Overview
Lichens are an essential component of global, national, and local biodiversity, but they can be difficult to identify to species level using morphology alone.
DNA barcoding is one approach to help improve identification of lichens. DNA barcoding is a method of using species-diagnostic regions of DNA known as DNA barcodes to characterise and identify organisms.
By extracting DNA, amplifying with PCR, and DNA sequencing DNA barcode regions, you can produce a DNA barcode that should be characteristic of that specimen. A specimen can then in theory (if not always in practice) be identified by using its DNA alone by comparison with reference databases of authenticated specimens, assuming that it has been previously barcoded.
The process of DNA barcoding lichens is very similar to that of other organisms, but there are some key differences involving:
- How to sample the material
- How to extract the DNA
- What primers to use to amplify the DNA for particular groups of lichens
- What can be expected with in terms of DNA barcode reference library completeness
This workflow will take you from sampling your lichen specimens, to DNA extraction, PCR, and examination of your PCR products before sending them off for DNA sequencing.
This resource was produced to support the British Lichen Society and their members in their future DNA barcoding work. We hope it will also be useful to lichenologists and nature lovers everywhere!